
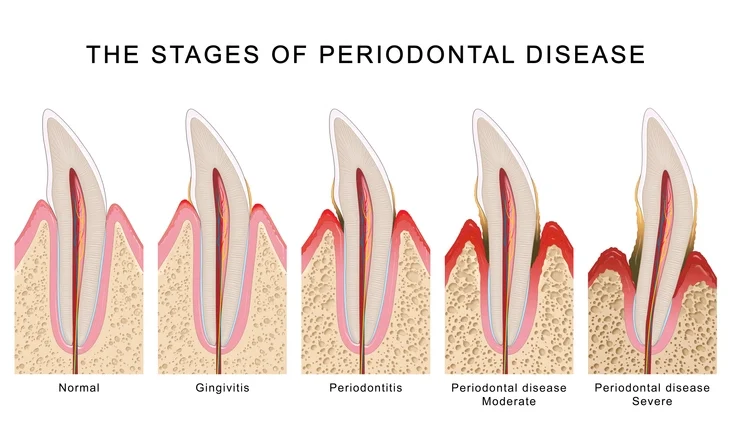
Periodontal disease represents one of the most prevalent chronic inflammatory conditions affecting humankind. It is characterized by the destruction of the supporting tissues of the teeth, including the gingiva, periodontal ligament, cementum, and alveolar bone. The primary etiological agent of periodontal disease is dental plaque, a complex biofilm composed of microorganisms embedded in an extracellular polymeric matrix. However, the onset and progression of the disease are multifactorial, involving intricate interactions between microbial biofilms and the host’s immune and inflammatory responses, modulated by environmental and genetic factors.
While gingivitis—an inflammation confined to the gingival tissues—is reversible, periodontitis involves irreversible loss of connective tissue attachment and alveolar bone. Understanding the aetiology of periodontal disease is essential for accurate diagnosis, prevention, and management.
Table of Contents
ToggleMicrobiological Basis of Periodontal Disease
1. The Role of Dental Plaque
Dental plaque is a structured microbial biofilm that adheres to the tooth surface, especially in the gingival crevice. Biofilms provide a protected environment that allows microorganisms to resist host defenses and antimicrobial agents. The biofilm’s architecture facilitates cell-to-cell communication (quorum sensing), exchange of genetic material, and coordinated metabolic activity, resulting in a highly resilient ecosystem.
Plaque formation begins with the pellicle, a thin layer of salivary glycoproteins and bacterial products that coats the enamel surface. Pioneer bacteria, primarily Streptococcus and Actinomyces species, colonize this pellicle. As the biofilm matures, secondary colonizers—such as Fusobacterium nucleatum—bridge early and late colonizers, leading to a complex community including anaerobic Gram-negative species like Porphyromonas gingivalis, Prevotella intermedia, and Tannerella forsythia.
The shift from a symbiotic to a dysbiotic community—one dominated by pathogenic species—marks the transition from health to disease.
2. Microbial Ecology and Biofilm Pathogenicity
The inflammatory response to plaque accumulation leads to gingival inflammation and the formation of a periodontal pocket, an anaerobic niche favorable to pathogenic microorganisms. Within this environment, microbial diversity increases, with a predominance of anaerobic and proteolytic species.
The red complex, described by Socransky et al. (1998), comprises three key pathogens strongly associated with chronic periodontitis:
- Porphyromonas gingivalis
- Tannerella forsythia
- Treponema denticola
Other associated species include Aggregatibacter actinomycetemcomitans (particularly in localized aggressive periodontitis), Fusobacterium nucleatum, Campylobacter rectus, Eikenella corrodens, and Prevotella intermedia.
Aggregatibacter actinomycetemcomitans is of particular importance in aggressive forms of periodontitis due to its potent leukotoxin and ability to invade epithelial cells, thereby evading host defenses.
Recent studies have also implicated viral pathogens—such as human cytomegalovirus (HCMV) and Epstein–Barr virus (EBV)—in modulating bacterial virulence and suppressing host immunity, thus contributing to the pathogenesis of periodontal lesions.
Theories on the Role of Plaque in Aetiology
The relationship between plaque and periodontal disease has been explained through several hypotheses that have evolved with advancing microbiological understanding. These hypotheses reflect the progression from a simplistic view of uniform microbial pathogenicity to a more nuanced model recognizing ecological and host–microbial interactions.
1. Non-Specific Plaque Hypothesis
The non-specific plaque hypothesis (NSPH) postulates that disease results from the overall quantity of plaque and the collective activity of the entire microbial community rather than specific pathogens. The severity of periodontal destruction is thus proportional to the amount of plaque present.
This concept emerged during the early 20th century, when the focus was on mechanical removal of plaque as the principal therapeutic goal. However, NSPH failed to explain why certain individuals develop severe periodontitis despite similar levels of plaque accumulation compared to those who remain healthy. Consequently, this hypothesis was replaced by more refined theories emphasizing qualitative microbial differences.
2. Specific Plaque Hypothesis
The specific plaque hypothesis (SPH), introduced by Loesche in the 1970s, posits that only a subset of microorganisms within the biofilm are responsible for disease initiation and progression. According to SPH, periodontopathogens such as P. gingivalis, A. actinomycetemcomitans, and T. forsythia possess virulence factors that enable them to disrupt host defenses and cause tissue destruction.
This hypothesis provided the rationale for antimicrobial therapy and diagnostic identification of key pathogens. However, it could not fully account for the complex microbial shifts seen in periodontitis or for disease occurrence in the absence of these specific organisms.
3. Ecological Plaque Hypothesis
Proposed by Marsh in 1994, the ecological plaque hypothesis (EPH) integrates the NSPH and SPH models. It suggests that disease arises from an ecological imbalance within the biofilm—dysbiosis—caused by environmental changes in the gingival crevice. Factors such as inflammation, nutrient availability (e.g., increased gingival crevicular fluid proteins), and redox potential favor the overgrowth of pathogenic species.
The EPH emphasizes that the host response contributes to this ecological shift: inflammation alters the environment, thereby promoting the proliferation of anaerobes and proteolytic bacteria. Consequently, therapeutic strategies should aim not only to reduce bacterial load but also to modify the ecological environment to restore symbiosis.
4. Keystone Pathogen Hypothesis
A more recent concept, the keystone pathogen hypothesis, proposed by Hajishengallis and colleagues, identifies specific pathogens (notably P. gingivalis) as key modulators of the host–microbe relationship. Despite being present in low abundance, these pathogens can disrupt immune homeostasis and alter the composition of the microbial community, thereby promoting dysbiosis and inflammation.
P. gingivalis exemplifies a keystone pathogen due to its capacity to manipulate complement pathways, inhibit neutrophil killing, and modulate Toll-like receptor signaling. These actions enable the survival of both itself and other commensal bacteria in an inflammatory environment, perpetuating tissue destruction.
Virulence Factors of Periodontal Pathogens
Pathogens employ multiple virulence mechanisms to invade tissues, evade immune responses, and cause destruction of periodontal structures. These mechanisms can be broadly categorized as adherence factors, invasion mechanisms, toxins and enzymes, and immune modulators.
1. Adherence and Colonization
Bacteria adhere to host tissues and other microbial cells using fimbriae, adhesins, and outer membrane proteins. For example, P. gingivalis expresses fimbriae (FimA proteins) that facilitate adhesion to epithelial cells and coaggregation with other species like Streptococcus gordonii.
2. Enzymatic Tissue Destruction
Periodontopathogens produce proteolytic enzymes such as collagenases, gelatinases, and trypsin-like proteases (notably gingipains in P. gingivalis). These enzymes degrade extracellular matrix components, immunoglobulins, and complement proteins, weakening the host barrier and promoting tissue breakdown.
3. Endotoxins and Exotoxins
Lipopolysaccharides (LPS) from Gram-negative bacteria stimulate macrophages and fibroblasts to release pro-inflammatory cytokines (IL-1β, TNF-α, and IL-6), prostaglandins, and matrix metalloproteinases (MMPs), all of which contribute to connective tissue destruction and bone resorption.
Aggregatibacter actinomycetemcomitans produces a potent leukotoxin that kills neutrophils and macrophages, impairing host defense mechanisms.
4. Modulation of Host Immune Response
Pathogens can subvert the host immune system to promote their persistence. For instance, P. gingivalis manipulates the complement system through its gingipains, leading to dysregulated inflammation and impaired clearance of microbes. Similarly, T. forsythia and T. denticola produce factors that inhibit neutrophil chemotaxis and oxidative burst.
Host Response in Periodontal Disease
The host response to microbial challenge determines the extent of tissue damage. Periodontal destruction is not caused directly by bacteria, but by the host’s inflammatory and immune response to the microbial biofilm. This response involves a complex interplay between innate and adaptive immunity.
1. Innate Immune Defenses
The innate immune system provides the first line of defense against microbial invasion.
Epithelial Barrier Function
The junctional epithelium forms a physical barrier, preventing bacterial penetration. It is rich in cell adhesion molecules and is capable of producing antimicrobial peptides such as β-defensins and cathelicidins. However, in disease, the junctional epithelium proliferates apically, forming a pocket epithelium that is more permeable and less protective.
Gingival Crevicular Fluid (GCF)
GCF contains plasma-derived proteins, antibodies, complement components, and inflammatory mediators. It flushes out non-adherent bacteria and delivers immune effectors to the site of infection.
Neutrophils
Neutrophils are the predominant leukocytes in the gingival sulcus. They migrate in response to chemotactic factors (IL-8, complement fragments C5a) and kill bacteria via phagocytosis, oxidative burst, and release of antimicrobial peptides. Defects in neutrophil function are strongly associated with aggressive periodontitis.
Macrophages and Dendritic Cells
These cells act as antigen-presenting cells (APCs), secreting cytokines that orchestrate inflammation. Macrophages release IL-1β, TNF-α, and prostaglandin E₂, mediating connective tissue degradation and osteoclast activation.
2. Adaptive Immune Response
The adaptive immune system provides specificity and memory. It includes humoral and cell-mediated immunity.
Humoral Response
Activated B cells differentiate into plasma cells that produce specific antibodies (IgG, IgA, IgM). These antibodies neutralize toxins and facilitate opsonization. However, in chronic periodontitis, antibody production may paradoxically enhance tissue destruction by forming immune complexes and perpetuating inflammation.
Cell-Mediated Response
T-helper (CD4+) cells play a central regulatory role. Th1 cells promote macrophage activation, while Th2 cells stimulate antibody production. More recently, Th17 cells have been implicated in periodontitis through their secretion of IL-17, which recruits neutrophils and enhances osteoclastogenesis.
Cytotoxic T cells (CD8+) and natural killer (NK) cells also contribute to host defense but may exacerbate tissue damage through cytotoxic mechanisms.
Cytokine Networks and Inflammatory Mediators
Cytokines are key regulators of periodontal inflammation. The balance between pro-inflammatory and anti-inflammatory cytokines determines disease progression or resolution.
- Pro-inflammatory cytokines: IL-1β, TNF-α, IL-6, and IL-17 stimulate MMP production and bone resorption.
- Anti-inflammatory cytokines: IL-10 and TGF-β counteract inflammation and promote healing.
- Prostaglandins (particularly PGE₂): promote vasodilation and osteoclastic bone resorption.
- Matrix metalloproteinases (MMPs): degrade collagen and extracellular matrix components.
The sustained overexpression of pro-inflammatory mediators results in a self-perpetuating cycle of tissue destruction and microbial proliferation.
Systemic and Environmental Modifying Factors
The host response and susceptibility to periodontal disease are influenced by several systemic and environmental factors.
1. Genetic Predisposition
Polymorphisms in genes encoding cytokines (e.g., IL-1, TNF-α) and Fc receptors have been associated with increased susceptibility to periodontitis.
2. Smoking
Tobacco use is a major risk factor, impairing neutrophil function, reducing blood flow, and altering microbial composition.
3. Systemic Diseases
Conditions such as diabetes mellitus amplify inflammatory responses and impair wound healing, thereby accelerating periodontal destruction.
4. Hormonal Changes
Pregnancy, puberty, and menopause can modify vascular permeability and immune responses, increasing susceptibility to gingival inflammation.
5. Stress and Nutrition
Psychological stress and nutritional deficiencies (e.g., vitamin C, D) may alter immune competence and affect periodontal health.
Clinical and Therapeutic Implications
Understanding the multifactorial aetiology of periodontal disease has significant clinical implications. Effective management requires a multifaceted approach targeting both microbial and host factors:
- Mechanical plaque control: Scaling and root planing to disrupt the biofilm.
- Antimicrobial therapy: Use of systemic or local antibiotics to target specific pathogens.
- Host modulation therapy: Agents such as doxycycline at sub-antimicrobial doses inhibit MMPs and modulate inflammation.
- Immunotherapy and probiotics: Emerging strategies aim to restore microbial balance and immune homeostasis.
Adjunctive measures—such as smoking cessation, glycemic control, and nutritional support—are essential for optimizing treatment outcomes.
Conclusion
Periodontal disease is a complex, multifactorial condition arising from the dynamic interplay between microbial biofilms and the host immune response. The evolution from the non-specific to ecological and keystone pathogen hypotheses reflects an increasing appreciation of this complexity. While bacterial infection initiates the disease, the destructive consequences are primarily host-mediated.
Future research focuses on the molecular mechanisms underlying host–microbe interactions, identification of microbial signatures predictive of disease, and development of targeted therapies to restore symbiosis. A comprehensive understanding of periodontal aetiology not only guides clinical management but also provides insights into systemic inflammatory diseases, underscoring the interconnection between oral and overall health.
References
- Hajishengallis, G., & Lamont, R. J. (2012). Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Molecular Oral Microbiology, 27(6), 409–419.
- Marsh, P. D. (1994). Microbial ecology of dental plaque and its significance in health and disease. Advances in Dental Research, 8(2), 263–271.
- Loesche, W. J. (1979). Clinical and microbiological aspects of chemotherapeutic agents used according to the specific plaque hypothesis. Journal of Dental Research, 58(12 Suppl), 2404–2412.
- Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C., & Kent, R. L. (1998). Microbial complexes in subgingival plaque. Journal of Clinical Periodontology, 25(2), 134–144.
- Slots, J. (2005). Herpesviral–bacterial interactions in periodontal diseases. Periodontology 2000, 38(1), 33–62.
- Page, R. C., & Kornman, K. S. (1997). The pathogenesis of human periodontitis: An introduction. Periodontology 2000, 14(1), 9–11.
- Seymour, G. J., Ford, P. J., Cullinan, M. P., Leishman, S., & Yamazaki, K. (2007). Relationship between periodontal infections and systemic disease. Clinical Microbiology and Infection, 13(S4), 3–10.
- Bartold, P. M., & Van Dyke, T. E. (2013). Periodontitis: A host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontology 2000, 62(1), 203–217.
- Tonetti, M. S., & Van Dyke, T. E. (2013). Periodontitis and atherosclerotic cardiovascular disease: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. Journal of Periodontology, 84(4 Suppl), S24–S29.
- Pihlstrom, B. L., Michalowicz, B. S., & Johnson, N. W. (2005). Periodontal diseases. The Lancet, 366(9499), 1809–1820.
- Kinane, D. F., Stathopoulou, P. G., & Papapanou, P. N. (2017). Periodontal diseases. Nature Reviews Disease Primers, 3(1), 1–14.
- Darveau, R. P. (2010). Periodontitis: A polymicrobial disruption of host homeostasis. Nature Reviews Microbiology, 8(7), 481–490.
- Chapple, I. L. C., & Genco, R. J. (2013). Diabetes and periodontal diseases: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. Journal of Clinical Periodontology, 40(S14), S106–S112.
- Curtis, M. A., Diaz, P. I., & Van Dyke, T. E. (2020). The role of the microbiota in periodontal disease. Periodontology 2000, 83(1), 14–25.
- Kinane, D. F., & Hart, T. C. (2003). Genes and gene polymorphisms associated with periodontal disease. Critical Reviews in Oral Biology & Medicine, 14(6), 430–449.